| Title of course: | Bioinformatics Algorithms (Module 2 of Fundamental Algorithms for Bioinformatics) |
| Lecturer: | Zsuzsanna Lipták |
| email: |
zsuzsanna DOT liptak AT univr DOT it |
| Course times: | Tue 12.30 - 14.30 (aula L) (we begin at 12:45) Thu 11.30 - 14.30 (aula A) (we take a small lunch break) |
| Student hours: |
Wed 9.30-11.30 and by appointment |
| Office: | Ca' Vignal, 2, 1st floor, right corridor, office 1.79 |
News:
GOALS of the course: To learn about some of the basic problems and algorithms behind common bioinformatics applications (sequence alignment, sequence similarity, sequence assembly, phylogenetics).
CREDITS: 12 CFU, together with module 1 of this course (Algorithm Design). This module contributes half of the grade. It is gained via a written exam, followed by oral exam. You are admitted to the oral exam only if you passed the written exam.
If you are taking this course as a student of Molecular and Medical Biotechnology (LM9) ("mutuazione Algorithms for Computational Biology"), then you will get 6 CFU (credits) for this course, and will have a different exam to take. However, it still holds that you have to take a written and an oral exam.
MATERIAL: When there are slides, I hand them out and put them up on the webpage. However, we always do more in class than what is on the slides: examples, missing proofs, additional material, illustrations, applications, etc. So you will always also need your class notes (or someone else's).
HOMEWORK: I regularly give you homework, and I expect you to do it. If you do not, you will not gain the experience in solving exercises that you need for the exam.
SYLLABUS:
Here is an overview of the topics that will be covered. The topics and the order may vary slightly.
SUGGESTED BOOKS:
Disclaimer: Here I list the topics which we covered in the lectures, include the slides, pointers to the respective chapters in books, and possibly other material of interest. However, remember that the slides are not enough for studying! You always need your class notes and exercises and/or books.
When I include corrections of slides, this refers to the handed out version. The slides which you can download here have already been corrected. If you find any further mistakes, please let me know!
| No. | Date | Topic | Slides (if any) | Homework (if any) | Supporting material or chapters in books, comments |
|---|---|---|---|---|---|
| 1 | Thu, 7-3-2019 | Organisation of course. Introduction: Strings and sequences in Biology and in Computer Science. | Organisation (printable), Strings and Sequences in Biology (printable), Strings and Sequences in Computer Science (printable) |
Give lower and upper bounds for the number of distinct prefixes, suffixes, substrings, and subsequences for a string s of length n over alphabet Sigma. | |
| 2 | Tue, 12-3-2019 | Pairwise alignment I: Number of alignments: recursion, explicit exponential lower bound, exhaustive search. Needleman-Wunsch DP-algorithm for global alignment, analysis. | Pairwise Alignment 1 (printable: part 1, part 2, part 3) | 1. Find all optimal global alignments of s=ACCT and t=CACT, using (a) match=2, mismatch=-1, gap=-1, and (b) match=1, mismatch=-1, gap=-2 2. Find all optimal local alignments between s=CATAC and t=AAGA, using using (a) match=2, mismatch=-1, gap=-1, and (b) match=2, mismatch=-2, gap=-1 |
This and the following lectures follow pretty closely chapter 3 of the Setubal & Meidanis book. (The Ohlebusch book and the Bielefeld Lecture Notes on Sequence Analysis both use distance functions instead of similariy functions for their presentation of alignment algorithms.) Number of alignments: Bielefeld Lecture Notes on Sequence Analysis (see above), appendix B. Correction of slides: on p. 24, the entry D(3,3) should be -1 (and not 1). |
| 3 | Thu, 14-3-2019 | Pairwise alignment II: Backtracing without and with traceback pointers. Space-saving variant of NW-algorithm. Local alignment: Smith-Waterman algorithm. Semiglobal alignment. | Pairwise Alignment 2 (printable: part 1, part 2) | Find the best approximate overlap(s) between s=ATCAT and t=GATAC using the appropriate version(s) of the semiglobal alignment algorithm with (a) match=1, mismatch=-1, gap=-1, and (b) match=1, mismatch=-1, gap=-2. | global al.: Setubal & Meidanis, ch. 3.2.1, local al.: ch. 3.2.2, semiglobal al.: ch. 3.2.3 Jones & Pevzner: global al: ch. 6.6, local al: ch. 6.8 |
| 4 | Tue, 19-3-2019 | Pairwise Alignment III: Affine gap functions in quadratic time and space. | same as before | (1) Compute the 3 matrices A,B,C for the strings s=AC and t=GA, using match=2, mismatch=-1, and gap: h=-3, g=-1. (2) same scoring function, with s=ACAGA, t=CAA |
affine gap functions: Setubal & Meidanis, ch. 3.3.3; Jones & Pevzner, ch. 6.9 Correction of slides: p. 10: "The problem now is that in cases 2. and 3. ..." (not: "cases 1. and 3.") |
| 5 | Tue, 26-3-2019 | Pairwise Alignment IV: Computing an optimal pairwise alignment in linear space via Divide-and-Conquer. | Pairwise Alignment 3 (printable) | In the lecture we used priority "left-diag-top". Repeat the computation with priority "top-diag-left" on the same example. | D&C algorithm for optimal al. in linear space: Bielefeld Lecture Notes on Sequence Analysis, ch. 5.4.; (a different algorithm for optimal al. in linear space: Setubal & Meidanis, ch. 3.3.1, and Ohlebusch ch. 8.1.2) Comment on the slides: The previous version of the slides ''PW al. 3'' is also correct, but this new version (the second one I handed out) is more detailed. Correction of slides: There was a mistake on p. 11: the entry (5,2) of the M-matrix is 0 (and not 2). p. 15: the sum should run from k=0 (not from k=1). |
| 6 | Thu, 28-3-2019 | Scoring matrices. | Scoring Matrices (printable) | Setubal & Meidanis, ch. 3.5.1 Here are two PAM mutation matrices, and two PAM scoring matrices (the ones handed out in the lecture). You can read the original article on PAM matrices (more exactly, book chapter), by Margret Dayhoff et al, 1978. It is very understandably written! Here is the BLOSUM62 matrix. See also this nice little article by Sean Eddy about the BLOSUM matrices (S. Eddy, Where did the BLOSUM62 alignment score matrix come from? Nature Biotechnology 22, pp. 1035-1036, 2004). |
|
| 7 | Tue, 2-4-2019 | Dotplots; Database search with BLAST. | Pairwise alignment in practice (printable) | Here is the orginal article on BLAST: S.F. Altschul et al. Basic Local Alignment Search Tool, J. Mol. Biol. 215: 403-410 (1990); and the one on BLAST2: S.F. Altschul et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs, Nucleic Acids Research, Vol. 25, No. 17: 3389-3402 (1997). There is a very detailed section on BLAST in the book by Mount (full ref. see above), within chapter 7, p. 300 onwards. BLAST is also described in Chapter 6.3 of the Bielefeld Lecture Notes on Seq. Analysis. Correction of slides: On p. 12, it should read "for DNA: w=11, for protein: w=3" (and not the other way around). |
|
| 8 | Thu, 4-4-2019 | BLAST (cont.); String distances 1: similarity vs. distance, Hamming distance, edit distance, DP algorithm for computing the edit distance, connection between alignment score and edit distance. |
String Distance 1 (printable) | (1) Compute the edit distance of s=TACAT and t=TGATAT (using unit cost edit distance), and give all optimal series of edit operations (both written as alignments, and in the form TACAT -> ... -> TGATAT). (2) Prove that the Hamming distance is a metric. |
String distances: Bielefeld Lecture Notes Seq. Analysis, ch. 3; similarity vs. distance: Setubal & Meidanis, ch. 3.6, Ohlebusch ch. 8.1.4 and 8.1.5;
edit distance and alignments: Jones & Pevzner ch. 6.4. Here is the original article by Vladimir Levenshtein: Binary codes capable of correcting deletions, insertions, and reversals, Doklady Akademii Nauk SSSR, 163 (4): 845-848 (1966). |
| 8a | Thu, 4-4-2019 | Extra lecture for biotechnologists: Computational efficiency. | Computational Efficiency 1 (printable), Computational Efficiency 2 (printable) | ||
| 9 | Tue, 9-4-2019 | String distances 2: LCS distance, q-gram distance: q-gram profiles, q-gram lemma, computation in linear time. | q-gram distance (printable) | (1) Show that the LCS-distance is a metric. (2) Develop a DP-algorithm for computing LCS(s,t). (3) Compute the q-gram distance of s=ACATTA and t=GACAT for q=3, using the sliding window algorithm from the lecture (use sigma=4). | q-gram distance: Bielefeld Lecture Notes on Sequence Analysis, ch. 3.7; Here is the article on QUASAR, a database search tool using q-grams: Stefan Burkhardt et al.: q-gram Based Database Searching Using a Suffix Array (QUASAR), RECOMB 1999: 77-83. Correction of slides: I have added the full example for the q-gram computation (new slide 15). On p. 14 what you see is the state of the array d_q after execution of line 2. |
| 10 | Thu, 11-4-2019 | de Bruijn sequences and (full) de Bruijn graphs: definitions, existence of dB-sequences, construction via dB-graphs. | (1) Find a dB-sequence of order 2 over Sigma={a,b,c}, and one of order 3 over {a,b}, using the appropriate de Bruijn graphs. (We did both of these in the lecture, so redo the graphs from scratch, without looking at your notes.) (2) Extra: Find a dB-sequence of order 3 over Sigma={a,b,c}, and one of order 4 over Sigma={a,b}. |
class notes and Compeau et al. article (see next lecture). | |
| 11 | Tue, 16-4-2019 | Fragment assembly with de Bruijn graphs I: Recap of SCS / overlap graph approach for Fragment Assembly, Eulerian graphs, Euler tours / Euler paths in de Bruijn graphs. | Fragment Assembly (printable) | Construct dB(s,3) for s=GATACATTTAT. Is there a string t different from s with 3-gram distance 0 to s? | See this article: How to apply de Bruijn graphs to genome assembly by Phillip E.C. Compeau, Pavel A. Pevzner, and Glenn Tesler, Nature Biotechnology, vol. 29 no. 11 (Nov. 2011); there is also supplementary material for this article. |
| 12 | Thu, 18-4-2019 | Fragment assembly with de Bruijn graphs II: dealing with errors, SBH (sketch). | same as before | Apply the de Bruijn graph approach to the fragment set F = {AGGTA, GGATGA, ATGAGG}, with q=3. | |
| 13 | Tue, 7-5-2019 | Text indexes: suffix trees. Simple construction of suffix trees (simple suffix insertion algorithm). | Suffix Trees 1 (printable) | Construct the ST of T=ANANAS using the Simple Suffix Insertion Algorithm. | Correction of slides: p. 6: Analysis (for p.m.); time for all-occurrences is O(k+occ_P). I also added two slides on suffix arrays. |
| 14 | Thu, 9-5-2019 | Discussion of previous exercises. Suffix trees: memory requirement, proof of linear storage space. | same as before | Bielefeld Lecture Notes on Sequence Analysis, ch. 7, contains more information about Suffix Trees. | |
| 15 | Tue, 14-5-2019 | Pattern matching on suffix trees, different variants of the problem. | Suffix Trees 2 (printable) | Construct the ST of the string MISSISSIPPI, using either of the two algorithms. Find all occurrences of ISSI, ISS, SISSI, and PISSI, using the ST. | |
| 16 | Thu, 16-5-2019 | Pattern matching on ST cont. Pre- and postorder traversal of ST. Computation of stringdepth and number of leaves in subtree. | same as before | Compute the two functions g(u) and sd(u) for the ST of MISSISSIPPI, using the post-order resp. pre-order traversal seen in class. | |
| 17 | Tue, 28-5-2019 | q-gram distance computation with ST cont. Suffix trees summary. | |||
| 18 | Thu, 30-5-2019 | Phylogenetics introduction: definition, different types of phylogenetic trees, number of phylogenetics trees. | Phylogenetics 1 (printable) | Bielefeld Phylogenetics Lecture Notes, ch. 1 and 2.4 | |
| 19 | Tue, 4-6-2019 (morning) | Distance based phylogenetic reconstruction: molecular clock, ultrametric matrices, additive matrices. | same as before | Bielefeld Phylogenetics Lecture Notes, ch. 7.1 | |
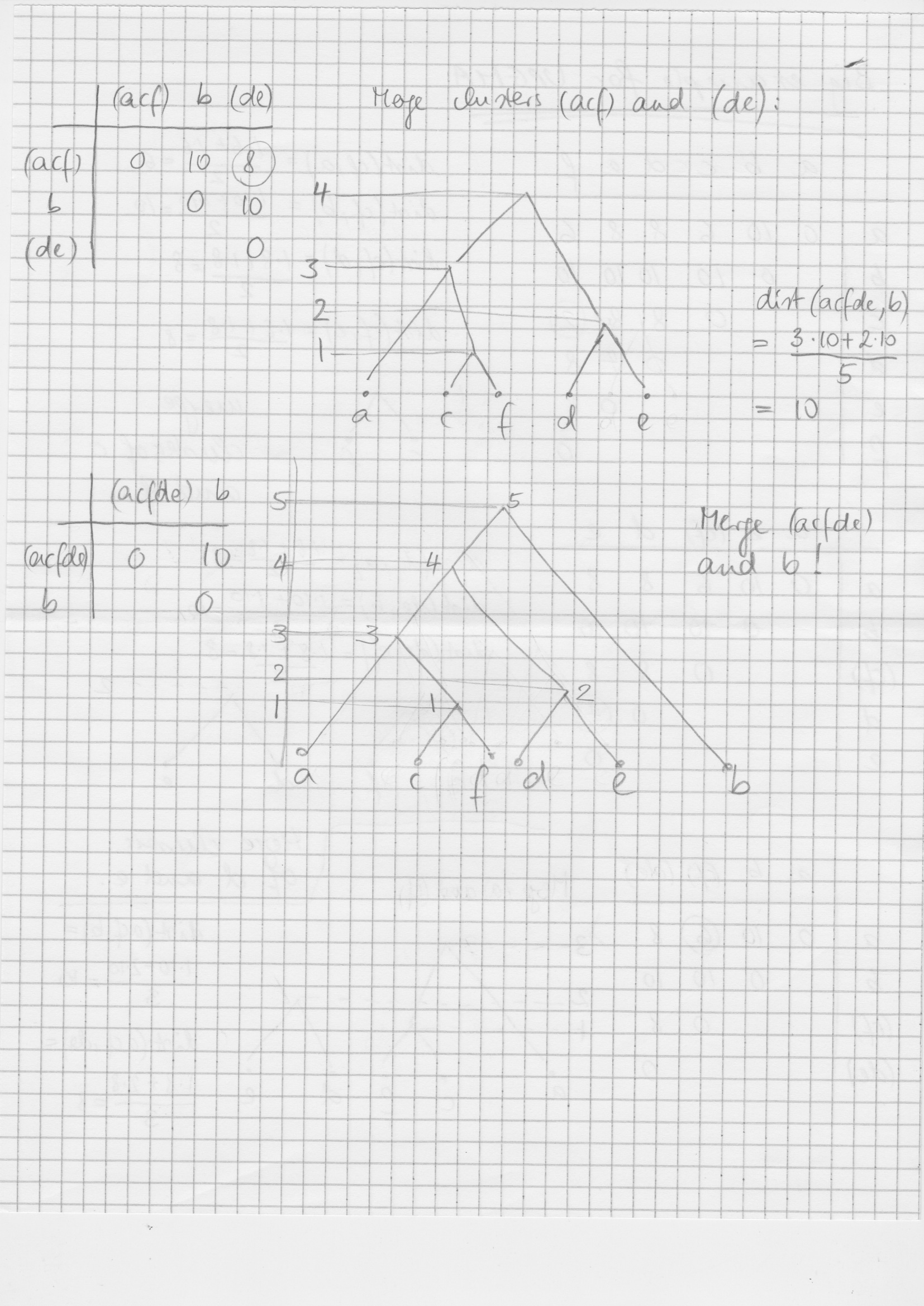
| 20 | Tue, 4-6-2019 (afternoon) | Distance based phylogenetic reconstruction: algorithm UPGMA. | same as before | Example on UPGMA: page 1 and page 2. | Bielefeld Phylogenetics Lecture Notes, ch. 7.2 |
| 21 | Thu, 6-6-2019 | Character-based phylogenetic reconstruction: homoplasies, compatibility. Perfect phylogeny. Parsimony, Large Parsimony vs. Small Parsimony, Fitch' algorithm. | Phylogenetics 2 (printable) | Example: Is this a PP? | Bielefeld Phylogenetics Lecture Notes, ch. 3 (characters and states), ch. 4 (Perfect Phylogeny, only p. 19 and 20), ch. 5.2 (Fitch' algorithm) |
| 22 | Fri, 7-6-2019 | Phylogenetic reconstruction in action: discussion of article about whale phylogeny. | Article on scientific controversy about whale phylogeny: Zhexi Luo: In search of the whales' sisters. Nature vol 404: p. 235-239, 2000. Here are two animations about whale evolution: from the Smithsonian Institute, Washington D.C.: animation, and a very sweet short one, from the Museum of New Zealand Te Papa Tongarewa: short animation. |
{kind=link}
{kind=link}